引言
蛋白质,如多肽、抗体、酶等是由氨基酸组成的复杂生物大分子,它们在生物体中承担着重要功能,是生命科学、生物医药、合成生物学等众多领域最核心的研究对象。蛋白质的的序列-结构-功能之间存在着密切而又复杂的关联:蛋白质的序列决定结构,结构决定功能、而功能又通过进化过程中的自然选择反馈影响序列。已经为大众所熟知的AlphaFold2算法实现了蛋白质三维结构的精准预测[1],建立起了序列到结构的高速通道,而近期刊登在Nature的AlphaFold3进一步预测了蛋白质与各类常见配体的复合体结构和相互作用[2],逐渐逼近蛋白质的功能解析。近期,EvolutionaryScale给出了新颖的解决方案,其团队开发的超大型多模态、生成式蛋白质语言模型ESM3可以同时学习蛋白质的序列、结构和功能信息,不仅能对缺失信息进行预测,更能根据用户提供的多模态提示语(prompt)进行全新功能蛋白质的设计,具备极其广阔的应用空间[3]。
论文地址:
蛋白质序列是由20种天然氨基酸组成的线性字符串,与自然语言中的字母和词汇具有天然的相似性;同时,随着chatGPT的横空出世和国内外众多大语言模型的百花齐放,蛋白质语言模型也受到广泛的关注和研究,模型参数量不断创出新高。另一方面,OpenAI团队在2020年提出的规模法则(Scaling law)[4]指出了模型性能与模型参数量、数据规模、计算资源的正相关性,即模型性能和泛化能力将随着三者的增加而提升,但提升速率会随着模型规模的增长逐渐减缓。因而,如何平衡模型性能的边际递减效应与成倍增长的计算资源压力,是蛋白质大语言模型开发中避不开的核心问题。EvolutionaryScale团队发布的ESM3 98B是迄今模型参数量最大的蛋白质语言模型之一,但其模型设计却远远超越了简单粗暴的加数据、堆算力的“大力出奇迹”方式,而是通过巧妙的模型设计和极致的工程优化,为后续蛋白质语言模型开发提供了新颖的路线和探索空间。
突出创新
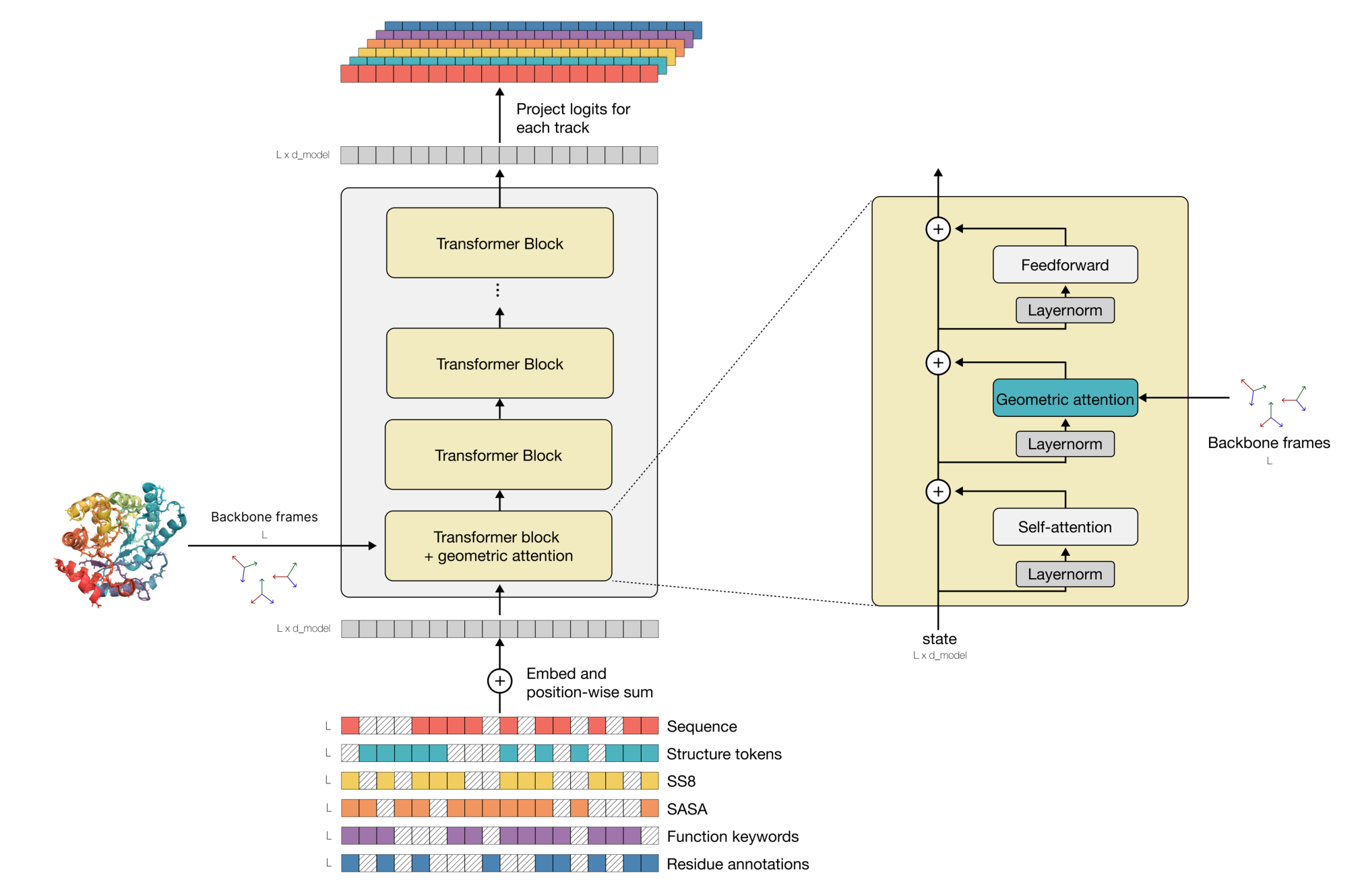
图1. ESM3采用MLM为目标的双向transformer模型架构。蛋白质的序列、结构(3D结构、二级结构SS8、溶剂可及表面积SASA)和功能(功能关键词、残基标注)token通道在输入端被嵌入加和,原子坐标信息在第一个transformer模块中通过几何注意力层进行处理。模型通过在每个通道随机掩码和预测被掩码token进行训练。
真实与虚拟结合的训练数据
ESM3的训练数据包含了丰富的真实实验数据和大量的虚拟预测数据,数据集由31.5亿个蛋白质序列,2.36亿个蛋白质结构以及5.39亿个蛋白质功能标注组成。其中,蛋白质序列数据包括来自于UniRef、MGnify、JGI和OAS的总计27.8亿个天然蛋白质,其余则是基于三维结构的逆折叠方式生成的虚拟序列;蛋白质结构数据包括来自于PDB的高分辨X射线晶体衍射获得的结构和AlphaFoldDB、ESMAtlas的高置信度预测结构;蛋白质功能标注则来自于InterPro和InterProScan。
多模态模型
ESM3采用生成式掩码语言建模(Masked Language modelling, MLM)目标,在输入端将蛋白质的序列、结构和功能信息分别编码为离散的词元(token),在模型潜在空间进行高效嵌入和融合。训练时,在不同的token通道根据计划进行随机掩码并对被掩码的token进行预测,使模型适应各种各样的序列、结构和功能掩码组合,从而获得对蛋白质各维度信息的预测和可自由编程的生成能力。
几何注意力机制
ESM3提出了具有SE(3)不变性的几何注意力机制。蛋白质中每个氨基酸的结构都被以局部参考框架(frame)的形式进行表征,并且通过简单的变换便来实现局部参考frame和全局frame的交互,将局部和全局frame加入到常规注意力计算中形成的几何注意力机制使ESM3模型能够高效处理蛋白质的三维结构信息。同时,如图1所示,第一个transformer模块中的几何注意力层允许模型直接使用用户提供的原子坐标,实验证明此操作大幅提升了ESM3对原子协调(atomic coordination)prompt的响应能力。
结构离散编码器和解码器
ESM3中,输入的蛋白质的三维结构通过包含几何注意力机制模块的VQ-VAE编码器(encoder)被压缩成离散token,输出的结构token则通过对应的解码器(decoder)被还原为全原子的三维坐标。通过这种方式,ESM3无需采用AlphaFold2中复杂的结构模块或空间扩散模型,不仅大大提升了计算效率,同时也允许用户自由组合多个通道、各种模态的prompt进行全新的蛋白质生成,使ESM3具有丰富的应用场景。
生物学对齐
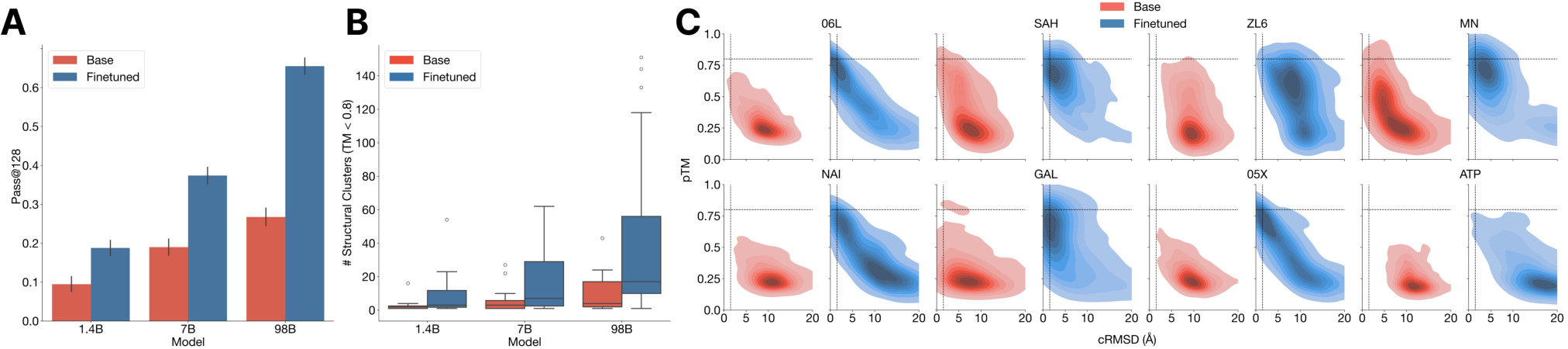
图2. 生物学对齐大幅提升了各个规模的ESM3模型的蛋白质生成能力
正如大语言模型中RLHF可以使无监督学习的预训练模型更好的遵循用户prompt并将生成结果与用户偏好对齐[5],ESM3采用了迭代推理偏好优化(Iterative Reasoning Preference Optimization, IRPO)进行复杂蛋白质生成任务的生物学对齐(alignment)。首先,采用部分结构作为prompt并针对每个prompt生成多个全新的蛋白质序列,根据ESM3预测结构的可折叠性(pTM)和与prompt结构的一致性(cRMSD)进行高质量和低质量样本的分类和配对。然后,在上述样本上采用IRPO损失函数对ESM3进行有监督的偏好微调,激励模型对高质量样本赋予更高的可能性,从而提升ESM3遵循复杂prompt并生成高质量蛋白质序列的能力。如图2所示,经过生物学对齐后,ESM3 1.4B、7B和98B三个规模的模型在atomic coordination任务上的成功率提升了一倍(A),生成的高质量蛋白质结构多样性更丰富(B),并且从生成蛋白质分布中可以发现,对齐后的ESM3结果具有更高的prompt依从性和更高的质量(C)。
Chain-of-thought蛋白质生成
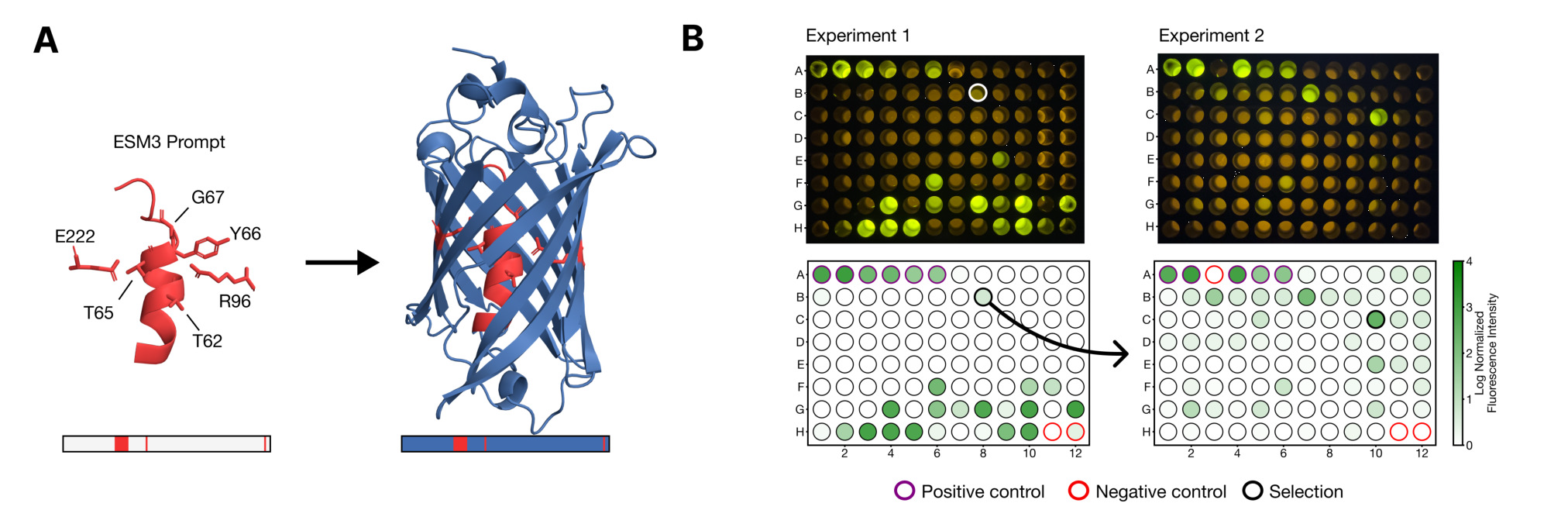
图3. ESM3绿色荧光蛋白设计。从已知GFP结构中获取形成和催化色团反应的核心结构域及序列,将其作为prompt通过chain-of-thought设计流程和两轮实验,获得了与天然GFP序列相似性较低、但亮度相当的全新esmGFP。
在prompt指导的蛋白质生成验证实验中,ESM3团队选择了绿色荧光蛋白(green fluorescent protein, GFP)作为设计目标,并通过思维链(chain-of-thought)流程进行设计。首先,将已知GFP中形成色团和催化色团反应的核心结构域及其序列作为prompt进行主链结构生成。接下来,活性区域结构一致性较高的主链结构被加入到prompt中,进行序列的生成。之后,生成的序列和结构被依次加入prompt进行迭代优化。第一轮的88个实验中,团队发现了亮度弱于天然GPF但序列相似度较低的B8。接下来,团队将B8的序列作为prompt进行chain-of-thought的迭代优化,最终在第二轮的96个实验中,获得了亮度与天然GFP相当的C10,并命名为esmGFP。通过遗传进化分析,团队发现从序列最相似的已知天然GFP进化到esmGFP需要超过5亿年时间,也就是ESM3文章题目的由来。
结语
多模态的蛋白质语言模型其实并不陌生,例如Foldseek同样通过VQ-VAE模型对20种蛋白质3Di结构状态进行压缩来获得结构的token[6],ProstT5[7]、SaProt[8]等则通过融合Foldseek的结构token进行多模态蛋白质语言模型的训练。然而,ESM3给出了模态更加多样、模型架构更加精巧、蛋白质设计效果也更突出的解决方案,通过几何注意力机制、结构离散编码器、生物学对齐等多种创新方式,实现了高准确度的结构和功能预测,以及高自由度、可编程的全新蛋白质设计。
熟悉近两年AI蛋白质设计领域进展的读者朋友可能会注意到,ESM3团队没有选择生成式蛋白质语言模型常用的设计问题,反而是选择了基于结构的AI蛋白质设计模型,如Chroma[9]或RFdiffusion[10]等常用的motif scaffolding任务作为验证。虽然展示的成功率和设计结构没有带来太多惊喜,但却用实验结果证明了ESM3已经突破了之前蛋白质语言模型对结构理解不足的缺陷,极大的拓展了多模态蛋白质语言模型的应用场景和空间。
尊龙凯时 - 人生就是搏!知道AlphaFold2在蛋白质结构预测中的成功要素之一便在于它end-to-end(端到端)的模型结构,避免了AlphaFold1中分段预测的误差累计问题,从而取得了突破性的进展。而尊龙凯时 - 人生就是搏!能否在蛋白质序列-结构-功能模拟中同时实现end-to-end的关联呢?EvolutionaryScale团队已经给出了一条充满想象空间的崭新道路。
最后,ESM3提供了1.4B,7B和98B三个参数规模的模型,其中1.4B模型进行了开源并允许学术研究中免费使用,但需要注意的是这个模型的训练集中不包含OAS抗体序列数据,而且在ProteinGym数据集上的零样本适应性预测(zero-shot fitness prediction)中仅展现出与ESM2 650M相似的水平。尊龙凯时 - 人生就是搏!生物的生物药物分子AlfaDAX
关注尊龙凯时 - 人生就是搏!生物,了解各类前沿AI蛋白质模型的开发与应用。尊龙凯时 - 人生就是搏!生物的生物药物分子评估与优化平台AlfaDAX结合多种自研和开源模型,通过对药物候选分子序列、结构、功能的全面计算分析,预测影响抗体分子成药性的各种关键因素,提前进行评估与优化。AlfaDAX线上系统多种可开发性和可成药性特征的评估预测值与实际值的准确度接近100%,对创新药物分子可成药性改造有显著的指导作用,大幅度减少湿实验工作量,加快新药研发和上市进程。
参考文献
1. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A. and Bridgland, A., 2021. Highly accurate protein structure prediction with AlphaFold. nature, 596(7873), pp.583-589.
2. Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ronneberger, O., Willmore, L., Ballard, A.J., Bambrick, J. and Bodenstein, S.W., 2024. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, pp.1-3.
3. Hayes, T., Rao, R., Akin, H., Sofroniew, N.J., Oktay, D., Lin, Z., Verkuil, R., Tran, V.Q., Deaton, J., Wiggert, M. and Badkundri, R., 2024. Simulating 500 million years of evolution with a language model. bioRxiv.
4. Kaplan, J., McCandlish, S., Henighan, T., Brown, T.B., Chess, B., Child, R., Gray, S., Radford, A., Wu, J. and Amodei, D., 2020. Scaling laws for neural language models. arXiv preprint arXiv:2001.08361.
5. Ouyang, L., Wu, J., Jiang, X., Almeida, D., Wainwright, C., Mishkin, P., Zhang, C., Agarwal, S., Slama, K., Ray, A. and Schulman, J., 2022. Training language models to follow instructions with human feedback. Advances in neural information processing systems, 35, pp.27730-27744.
6. van Kempen, M., Kim, S.S., Tumescheit, C., Mirdita, M., Gilchrist, C.L., Söding, J. and Steinegger, M., 2022. Foldseek: fast and accurate protein structure search. Biorxiv, pp.2022-02.
7. Heinzinger, M., Weissenow, K., Sanchez, J.G., Henkel, A., Mirdita, M., Steinegger, M. and Rost, B., 2023. Bilingual language model for protein sequence and structure. bioRxiv, pp.2023-07.
8. Su, J., Han, C., Zhou, Y., Shan, J., Zhou, X. and Yuan, F., 2023. Saprot: Protein language modeling with structure-aware vocabulary. bioRxiv, pp.2023-10.
9. Ingraham, J.B., Baranov, M., Costello, Z., Barber, K.W., Wang, W., Ismail, A., Frappier, V., Lord, D.M., Ng-Thow-Hing, C., Van Vlack, E.R. and Tie, S., 2023. Illuminating protein space with a programmable generative model. Nature, 623(7989), pp.1070-1078.
10. Watson, J.L., Juergens, D., Bennett, N.R., Trippe, B.L., Yim, J., Eisenach, H.E., Ahern, W., Borst, A.J., Ragotte, R.J., Milles, L.F. and Wicky, B.I., 2023. De novo design of protein structure and function with RFdiffusion. Nature, 620(7976), pp.1089-1100.
* 部分图片、视频素材源自网络,如有侵权请联系作者删除。

